
Pendant la pandémie de Covid, le gouvernement américain a dépensé des milliards de dollars pour près de 400 produits destinés à protéger, diagnostiquer et traiter des centaines de millions de personnes – tous portant le label « EUA » ou « Emergency Use Authorization ».
Mais que signifie réellement EUA ?
Avant même de répondre à cette question, et afin de comprendre où se situe l'EUA par rapport aux autres voies d'autorisation ou d'approbation des produits médicaux, il est utile d'examiner ce que l'EUA n'est pas:
L'EUA n'est pas une désignation pour un produit expérimental faisant l'objet d'un essai clinique
Si nous ne comprenons qu’une chose à propos de l’EUA, ce devrait être ceci : l’EUA ne s’applique pas à un produit faisant l’objet d’un essai clinique régi par les réglementations de la FDA ou d’autres exigences légales.
L’EUA n’est pas non plus la même chose que l’accès élargi (EAU), souvent appelé accès « à usage compassionnel », qui consiste à accorder aux patients atteints de maladies graves et incurables l’accès à des produits expérimentaux avant qu’ils ne soient entièrement approuvés.
Ce tableau d'un Présentation FDA-CDC 2020 résume les différences entre les produits faisant l’objet d’essais cliniques, les produits donnés aux patients grâce à un accès « compassionnel » élargi et les produits autorisés par l’EUA :
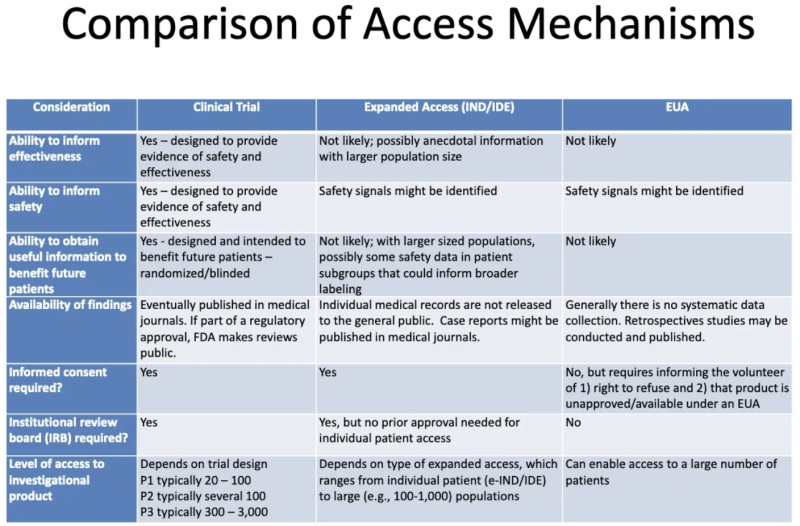
Voici ce que ce tableau nous dit sur l'EUA :
- Le processus d'octroi de l'EUA n'est pas susceptible de générer des informations sur l'efficacité d'un produit.
- Le processus d'octroi de l'EUA n'est pas conçu pour fournir des preuves de sécurité ou d'efficacité, mais des signaux de sécurité peuvent être identifiés.
- Il est peu probable qu’une fois qu’un produit a obtenu l’EUA et été administré à certains patients, des informations utiles soient obtenues au profit de futurs patients.
- Il n'existe aucune collecte systématique de données sur l'efficacité ou la sécurité de l'EUA, et aucune donnée n'est publiée dans des revues médicales dans le cadre du processus d'approbation réglementaire.
- Aucun consentement éclairé n'est requis, mais les patients qui se portent volontaires pour prendre le produit doivent être informés qu'ils peuvent refuser et que le produit n'est pas approuvé/disponible dans le cadre de l'EUA.
- Aucun comité d'examen institutionnel (CISR) n'est requis. [CISR est un conseil censé protéger le bien-être des sujets humains dans les essais cliniques]
Pour clarifier encore plus à quel point l'EUA est distinct de tout processus d'approbation normal, dans un Publication 2009 de l'Institut de médecine des académies nationales, on retrouve cette affirmation :
Il est important de reconnaître qu’une EUA ne fait pas partie du chemin du développement ; il s'agit d'une entité entièrement distincte qui n'est utilisée qu'en cas d'urgence et ne fait pas partie du processus d'approbation des médicaments. (p. 28)
Pour résumer:
Il est peu probable que le processus d’octroi de l’EUA à un produit génère des preuves de sécurité ou d’efficacité. Une fois qu'un produit a obtenu l'EUA et est administré aux patients, il est peu probable que des informations utiles soient obtenues pour bénéficier aux futurs patients, car il n'existe pas de collecte systématique de données sur l'efficacité ou la sécurité.
Sur la base de toutes ces informations très claires provenant du CDC/FDA et de l'IMNA, il serait juste de conclure que l'autorisation d'utilisation d'urgence est un processus qui doit être appliqué de manière très judicieuse et uniquement en cas d'urgence extrême.
Voyons maintenant à quels types de situations d'urgence l'EUA est légalement conçue pour répondre.
L'EAU est destinée aux urgences liées aux ADM
Les lois autorisant le « mécanisme d’accès » de l’EUA décrit ci-dessus ont été élaborées pour les cas d’urgences extrêmes et immédiates impliquant des armes de destruction massive (ADM), également appelées agents CBRN (chimiques, biologiques, radiologiques, nucléaires).
Voici comment la Food & Drug Administration (FDA) décrit ses pouvoirs EUA:
Article 564 de la loi FD&C (21 USC 360bbb-3) permet à la FDA de renforcer les protections de santé publique contre les agents biologiques, chimiques, nucléaires et radiologiques.
Grâce à cette autorité EUA, la FDA peut contribuer à garantir que des contre-mesures médicales peuvent être utilisées en cas d'urgence pour diagnostiquer, traiter ou prévenir des maladies ou affections graves ou potentiellement mortelles causées par des agents biologiques, chimiques, nucléaires ou radiologiques lorsqu'il n'existe pas de mesures adéquates et approuvées. , et les alternatives disponibles (entre autres critères).
Ces pouvoirs EUA ont été accordés en 2004 dans des circonstances très spécifiques liées à la préparation aux attaques d'agents CBRN.
Comme expliqué dans le Bill of Health de Harvard Law,
En fin de compte, c’est la guerre contre le terrorisme qui donnera lieu à une autorisation d’utilisation d’urgence. Après les événements du 11 septembre 2001 et les attaques ultérieures par courrier à l'anthrax, le Congrès a adopté la Loi sur le projet Bioshield de 2004.
Les record indique que le Congrès se concentrait spécifiquement sur la menace de bioterrorisme, et non sur la préparation à une pandémie naturelle.
Compte tenu d’un type aussi restreint de situations d’urgence véritablement extrêmes impliquant une attaque par ADM, il est compréhensible que le « mécanisme d’accès » de l’EUA n’exige pas beaucoup de surveillance réglementaire ou le respect de normes de fabrication ou d’essais cliniques.
Alors, que nécessite réellement le mécanisme d’accès à l’EUA ?
Les 3 étapes pour l’autorisation d’utilisation d’urgence (EUA)
Trois choses doivent se produire pour que l’EUA soit accordée à un produit médical :
- Le secrétaire à la Sécurité intérieure, le secrétaire à la Défense ou le secrétaire à la Santé et aux Services sociaux doivent déterminer s'il existe une urgence impliquant une attaque ou une menace d'attaque avec un agent CBRN ou une maladie causée par un tel agent.
- La FDA doit s’assurer qu’elle répond à quatre « critères statutaires » lorsqu’elle délivre l’EUA.
- La FDA doit « imposer certaines conditions requises » dans l’EUA.
EUA Étape 1 : Déclarer une urgence CBRN
La déclaration d'urgence pour l'EUA est distincte et sans rapport avec toute autre déclaration d'urgence qui peut être émise par le président, le secrétaire du HHS ou toute autre personne. Il doit être délivré spécifiquement dans le but d’activer l’EUA et peut être résilié ou prolongé indépendamment de toute autre déclaration d’urgence.
Voici ce que la loi EUA stipule Voici les quatre scénarios possibles pour activer le « mécanisme d’accès » de l’EUA :
- une détermination du secrétaire à la Sécurité intérieure selon laquelle il existe une urgence nationale, ou un potentiel important d'urgence nationale, impliquant un risque accru d'attaque avec un ou plusieurs agents biologiques, chimiques, radiologiques ou nucléaires ;
- une détermination du secrétaire à la Défense selon laquelle il existe une urgence militaire, ou un potentiel important d'urgence militaire, impliquant un risque accru pour Uni États les forces militaires, y compris le personnel opérant sous l'autorité du titre 10 ou du titre 50, d'attaquer avec :
- un ou plusieurs agents biologiques, chimiques, radiologiques ou nucléaires ; ou
- un ou plusieurs agents susceptibles de causer, ou qui sont autrement associés à, un risque spécifique et potentiellement mortel pour United États forces armées;
- une détermination par le Secrétaire [de la Santé et des Services sociaux] qu'il existe une urgence de santé publique, ou un potentiel important d'urgence de santé publique, qui affecte, ou est susceptible d'affecter de manière significative, la sécurité nationale ou la santé et la sécurité de Uni États les citoyens vivant à l’étranger et qui implique un ou plusieurs agents biologiques, chimiques, radiologiques ou nucléaires, ou une maladie ou une affection pouvant être imputable à ce ou ces agents ; ou
- l’identification d’une menace matérielle en application de l’article 319F-2 du Loi sur le service de santé publique [42 U.S.C. 247d-6b] suffisant pour affecter la sécurité nationale ou la santé et la sécurité des Uni États citoyens résidant à l’étranger.
EUA Étape 2. Répondre aux critères statutaires
Une fois que l’un des secrétaires a déclaré qu’il existe une urgence justifiant l’EUA, quatre autres « critères statutaires » doivent être remplis pour que la FDA puisse délivrer l’EUA. Voici comment la FDA explique ces exigences:
- Maladie ou affection grave ou potentiellement mortelle
Pour que la FDA délivre une EUA, le ou les agents CBRN mentionnés dans la déclaration EUA du secrétaire du HHS doivent être capables de provoquer une maladie ou un état grave ou potentiellement mortel.
- Preuve d'efficacité
Les produits médicaux qui peuvent être pris en compte pour une EUA sont ceux qui « peuvent être efficaces » pour prévenir, diagnostiquer ou traiter des maladies ou affections graves ou potentiellement mortelles qui peuvent être causées par un ou plusieurs agents CBRN identifiés dans la déclaration du secrétaire du HHS. urgence ou menace d’urgence en vertu de l’article 564(b).
La norme « peut être efficace » pour les EUA fournit un niveau de preuve inférieur à la norme « d'efficacité » utilisée par la FDA pour l'approbation des produits. La FDA a l'intention d'évaluer l'efficacité potentielle d'un éventuel produit EUA au cas par cas à l'aide d'une analyse risques-avantages, comme expliqué ci-dessous.
[FACE EN GRAS AJOUTÉ]
- Analyse risques-avantages
Un produit peut être pris en considération pour une EUA si le commissaire détermine que les avantages connus et potentiels du produit, lorsqu'il est utilisé pour diagnostiquer, prévenir ou traiter la maladie ou l'affection identifiée, l'emportent sur les risques connus et potentiels du produit.
Pour déterminer si les avantages connus et potentiels du produit l'emportent sur les risques connus et potentiels, la FDA a l'intention de regarder à l'ensemble des preuves scientifiques pour établir une détermination globale des risques et des avantages. Une telle preuve, qui pourrait survenir provenant de diverses sources, Peut inclure (sans s'y limiter) : résultats d'essais cliniques nationaux et étrangers, données d'efficacité in vivo provenant de modèles animaux et données in vitro, disponible pour examen par la FDA. La FDA évaluera également la qualité et la quantité des preuves disponibles, compte tenu de l’état actuel des connaissances scientifiques.
[FACE EN GRAS AJOUTÉ]
- Aucune alternative
Pour que la FDA délivre une EUA, il ne doit exister aucune alternative adéquate, approuvée et disponible au produit candidat pour diagnostiquer, prévenir ou traiter la maladie ou l'affection. Un produit alternatif potentiel peut être considéré comme « indisponible » si les stocks de l’alternative approuvée sont insuffisants pour répondre pleinement aux besoins d’urgence.
EUA Étape 3. Imposer les conditions requises
Une fois que nous avons la déclaration d’urgence spécifique à l’EUA, et une fois que la FDA a déterminé que le produit peut être efficace et que toutes les preuves disponibles montrent que ses avantages l’emportent sur ses risques, il existe un niveau supplémentaire de réglementation connexe.
Voici comment un Rapport 2018 du Congressional Research Service sur l'EUA explique ceci:
FFDCA §564 ordonne à la FDA d'imposer certaines conditions requises dans un EUA et autorise des conditions discrétionnaires supplémentaires, le cas échéant. Les conditions requises varient selon que l'EUA concerne un produit non approuvé ou une utilisation non approuvée d'un produit approuvé. Pour un produit non homologué, les conditions d’utilisation doivent :
1° s'assurer que les professionnels de la santé qui administrent le produit reçoivent l'information requise;
(2) s'assurer que les personnes à qui le produit est administré reçoivent les informations requises ;
(3) prévoir la surveillance et la déclaration des événements indésirables associés au produit ; et
(4) prévoir la tenue de registres et la production de rapports par le fabricant.
Conclusion
Comme indiqué dans cet article, la FDA/CDC reconnaissent clairement qu'il est peu probable que le processus d'octroi d'une autorisation d'utilisation d'urgence (EUA) génère des informations sur l'efficacité ou la sécurité d'un produit. Lorsque nous examinons la lettre de la loi régissant l’EUA, nous constatons qu’il s’agit effectivement d’une évaluation correcte.
La loi EUA n’impose aucune norme légale ou réglementaire susceptible de déterminer si un produit est sûr ou efficace. Les seules normes sont de savoir si la FDA estime que le produit peut être efficace et si ses avantages connus l'emportent sur ses inconvénients connus. S'il n'y a aucun préjudice ou avantage connu, parce que le produit n'a jamais été soumis au processus d'approbation du médicament, la FDA peut utiliser les informations ou les normes de son choix pour prendre cette décision.
Il résulte de tout cela qu'une entreprise dont le produit est candidat à l'EUA peut tenter de démontrer la sécurité et/ou l'efficacité du produit par tous les moyens de son choix. L’existence d’une telle tentative (qu’il s’agisse d’un essai clinique ou d’un autre mécanisme de collecte de données) et la manière dont cette tentative est menée dépendent de l’entreprise. Rien dans la loi EUA ne s’applique à la manière dont l’entreprise conçoit, mène ou analyse les études ou autres mécanismes de collecte de données qu’elle choisit de poursuivre.
Appliqué aux produits Covid, cela signifie :
- Aucune donnée de sécurité ou d’efficacité issue des essais cliniques n’était requise pour que les produits Covid reçoivent l’EUA.
- Tous les essais cliniques mentionnés dans le processus EUA ont été menés sans normes réglementaires légalement applicables.
- Quand on découvre que ces produits manquent d’efficacité ou de sécurité, ce n’est pas une surprise. C’est un résultat très probable du processus.
- Il n’existe aucune donnée issue du processus EUA sur laquelle fonder des décisions non-EUA concernant la sécurité ou l’efficacité du produit. Ainsi, toute utilisation non-EUA du produit nécessiterait dès le début de passer par le processus d’approbation légale pour les produits médicaux ordinaires.
En savoir plus sur le processus d’approbation des vaccins Covid ici.
Réédité de l'auteur Sous-empilement
Publié sous un Licence internationale Creative Commons Attribution 4.0
Pour les réimpressions, veuillez rétablir le lien canonique vers l'original Institut Brownstone Article et auteur.








